Major areas of interest at the present time include Medicinal Chemistry, Synthetic Organic Chemistry, and Natural Products Chemistry. More specifically, we are interested in the synthesis of natural products with biological activity or related analogs with enhanced activity. Research of this type has led us into the alkaloid, quinoline, indole, β-carboline, benzodiazepine and coumarin fields. Much of our research effort has been concerned with the synthesis of small molecules active in the CNS, to search for receptor subtype selective activity.
A. The benzodiazepines employed to treat anxiety are a group of compounds with wide therapeutic application as anxiolytics, anticonvulsants, hypnotics and muscle relaxants. However, the sedative-hypnotic, muscle-relaxant, ataxic and amnesic side effects limit their use in the elderly and in many other patients. Recently, a number of β-carbolines were prepared in our laboratory which have intrinsic effects opposite to the benzodiazepines (anxiogenic, convulsant, etc.) and have been employed to study mechanisms in anxiety. Moreover, there is some selectivity [Bz1-selective (Bz1 = α1β3γ2), BCCt and 3-PBC antagonists] in their mode of action in vivo. More importantly, we have recently discovered inverse agonist and antagonist activity in the rigid, planar pyridodiindole Bz receptor ligands which simplifies computer modeling of the receptor subtypes. A collaboration with a network of 19 pharmacologists has been set up to search for selective action, work in progress is detailed below:
- To prepare new and better anxioselective anxiolytics. These agents would be selective for antianxiety effects but would not possess sedative-hypnotic, ataxic or muscle-relaxant effects; a BzR selective agonist potentially devoid of abuse potential. We have now prepared anxioselective anxiolytics devoid of muscle-relaxant, ataxic and sedative-hypnotic effects. These orally active agents are currently being characterized and should be useful in treatment of panic disorders, general anxiety disorders, social anxiety disorders, agrophobias and post traumatic stress disorders. It appears potential abuse potential for these new agents is decreased in comparison to that of Valium, Xanax, etc.
- To prepare new and better sleep inducing agents than Dalmane. These would be compounds selective for sedative-hypnotic properties to the exclusion of side effects (BzR α1 selective ligands).
- To prepare potent, long-lived, selective (Bz1, Bz 2, Bz 5, Bz 6, etc) benzodiazepine receptor selective agonists and antagonist/inverse agonists. These inverse agonists/ antagonists would be employed to reverse the effects of benzodiazepine-induced anesthesia and to indirectly reverse barbiturate-induced CNS depression. This would help in surgery in the elderly, who oftentimes experience respiratory arrest with the barbiturate/haloalkanes presently in use.
- Study the effect of anxiogenic agents on the levels of endorphins in the brain (Endocrinology, 1985).
- Use the SAR of the rigid, planar pyridodiindoles to computer model the topography of each of the benzodiazepine receptor subtypes (Bz1-Bz6). This has been published, but 200 new ligands will be added to this modeling strategy this year.
- To differentiate between Bz1, Bz5, and Bz6 receptors as well as between Bz1, Bz2 and Bz3 receptor subtypes.
- To determine if the ability of Ro 15-4513 to reverse the effects of alcohol is mediated by the Bz6 [diazepam-insensitive site (Bz6)] site, the Bz5 site, the Bz1 receptor site or a related site. The α1 preferring, orally active BzR antagonists, BCCt and 3PBC have recently been shown to reverse alcohol self-administration in rats, and may have clinical potential in the treatment of alcoholism. These agents exhibit very weak anxiolytic activity in alcoholic rat models as well as potently reversing alcohol self-administration.
- To employ our Bz5 receptor ligands to design BzR ligands with 200 fold selectivity for Bz5 sites after which the pharmacology of the Bz5 site can be elucidated to use these to enhance cognition and to treat Alzheimer’s disease.
- To employ the same approach under 8 with molecular modeling and chemical synthesis to define the pharmacology of Bz1 and Bz6 subtypes.
- Eventually, with receptor subsite selective ligands, define the exact pharmacology of all major BzR (GABA(A) sites and employ this in drug design for the preparation of anxioselective anxiolytics, anticonvulsants and sedative hypnotics with decreased abuse potential and a better understanding of tolerance.
- Study the interrelationship between GABAergic and dopaminergic systems, in regard to the prevention of alcohol/drug abuse. This work is presently underway in collaboration with Dr. David Segal and Ron Kuczenski at UCSD via microdialysis techniques.
- Ultimate goal – to determine the differences at the molecular level between the various BzR sites via modeling, synthesis, site directed mutagenesis, cloning, and X-ray crystallography of irreversibly bound photolabels to the specific GABA(A)/BzR Cl ion channels, beginning with α1β3γ2 and α5β3γ2 subtypes.
- To use Bz5 (α5β3γ2) subtype selective inverse agonists and agonists to push forward our GABA approach for the treatment of Alzheimers disease and age-associated memory impairment in the elderly.
B. In mammals L-tryptophan and other indoleamines are oxidized to formylkynurenines via the kynurenine pathway by indoleamine 2,3-dioxygenase (IDO) or tryptophan 2,3-dioxygenase (TDO). Although both enzymes catalyze the same transformation, TDO is found only in the liver, while IDO is present in a wide variety of tissues such as brain, lung, and small intestine, as well as macrophages that are found in the CNS. Furthermore, IDO is a 4l kD, monomeric heme containing protein that utilizes superoxide to cleave the 2,3-double bond of indoleamines.
Many inflammatory diseases and neurodegenerative diseases have been hypothetically linked to aberrant L-tryptophan metabolism caused by activation of IDO. Moreover, Heyes and coworkers have recently reported evidence which implicates the activation of IDO in inflammatory diseases, such as AIDS, dementia and wasting, meningitis and sepsis. Interferon γ has been shown to induce the production of IDO. One of the ways in which the body responds to infection by foreign organisms and head injuries is to produce large amounts of interferon-gamma and other immune system activators. High levels of interferon-gamma eventually induce the production of high levels of IDO which results in the catabolism of large amounts of L-tryptophan and the production of high levels of metabolites of the kynurenine pathway. This can lead to extremely high levels of quinolinic acid in the CNS which can effect many neuropathological changes including nerve cell death and dementia.
Recently we have prepared both competitive and noncompetitive (modulatory β-carboline binding site) inhibitors of IDO (Med. Chem. Res., 1993, 1994). Na-methyl L(-)-tryptophan is the most active competitive inhibitor reported to date; however, the noncompetitive inhibitors 3-nitro β-carboline and 3-butyl β-carboline look even more promising. We plan, via molecular modeling and chemical synthesis, to search for even more potent inhibitors of IDO in collaboration with Dr. Melvin Heyes and Dr. Markey at NIH as well as Dr. Jamie (Australia). Drs. Mellor and Dunn have recently employed Na-methyl-D-tryptophan (prepared first in this laboratory) as an IDO inhibitor with clinical potential. The Na-methyl-D-tryptophan, first prepared and reported here in Milwaukee, has been employed by Drs. Mellor and Dunn to shed light on the “pregnancy paradox” and is being studied as a treatment for immune-system related diseases.
C. Multidrug resistance (MDR) in neoplastic cells is usually due to decreased cellular retention of drugs such as vincristine or doxorubicin. An ATP-dependent drug efflux pump has been detected in MDR-1-phenotypic cells and inhibition of the MDR pump is presumably a primary mechanism for reversal of MDR. Although quinine and quinidine are reversal agents and inhibitors of the MDR pump, a mixture of diastereomeric epoxides of quinine 10,11-epoxide was 8 fold more active in inhibiting the MDR pump. It is believed one of the diastereomers of the epoxide is covalently (Mol. Pharmacol., 1994) bonding to the pump. At present, the stereospecific synthesis of both epoxides is under study to determine which is the active agent, and how effective. Moreover, other agents known to inhibit this pump will be converted into epoxides to attempt to increase activity. Goal – find a way to block the pump so that vincristine/etc can kill drug resistant strains of cancer cells. The natural product tryprostatin-A, synthesized first here in Milwaukee, is the most potent inhibitor of breast cancer resistant protein reported to date (Joel Turner). Analogs of this natural product are being synthesized to find even more potent inhibitors of this member of the ABC transporter family. The goals of much of this work are to develop small molecules which can be employed to treat multidrug resistant forms of cancer.
D. The MDR pump is reported to be ubiquitous in nature, consequently, our pure diastereomeric quinine-10,11-epoxides will be employed with quinine and chloroquine in drug resistant strains of P.falciparum to determine if this simple approach would be effective in treating drug-resistant strains of malaria.
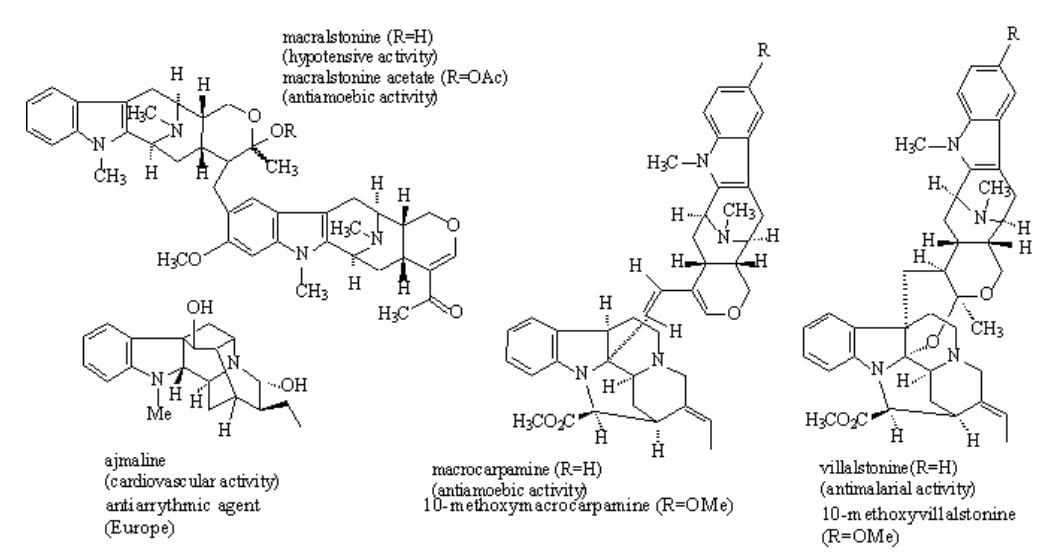
Figure 1
E. During recent years an increasing number of macroline related alkaloids has been isolated from various species of Alstonia. At the present time this group contains over 90 indole alkaloids, at least 21 of which are bisindoles. The hypotensive base macralstonine isolated from Alstonia macrophylla Wall is a member of this family, as well as the bisindole villalstonine. This latter alkaloid has been found to exhibit potent antimalarial activity against Plasmodia falciparum, while the related macrocarpamine is active against Entamoeba histolytica and Plasmodia falciparum. One of these alkaloids is active against drug resistant strains of Plasmodia falciparum while the other is active only at the chloroquine sensitive strain. The activity of these alkaloids confirms the use of Alstonia angustifolia in traditional medicine for the treatment of malaria as well as amoebic dysentery.

Figure 2
The enantiospecific total synthesis of these bisindole alkaloids (see Figures 1 and 2) as well as the cardiovascularly active alkaloid ajmaline and a number of other macroline related sarpagine alkaloids is being pursued. The synthetic route to these bisindole alkaloids which employs the trans, stereospecific Pictet-Spengler reaction developed in this laboratory is both enantiospecific and doubly convergent [J. Am. Chem. Soc. (1994); J. Org. Chem. (2003)]. Recently the total synthesis of the bisindoles macralstonine and macralstonidine [J. Org. Chem. (2003)] as well as the partial synthesis of macrocarpamine and villalstonine has been completed and reported. A number of indole alkaloids (over 30) have been synthesized over the past three years employing the recently developed enolate-driven palladium-catalyzed cross coupling reaction in combination with the asymmetric Pictet-Spengler reaction. A related palladium-mediated method of Larock has been employed to develop the first stereospecific, regiospecific synthesis of 6-methoxy and 7-methoxy (D or L) tryptophan building blocks for total synthesis or as IDO active ligands.
F. Attempts to prepare strained 10pi and 14pi annulenes (see below) termed cyclopentapentalenes in order to study homoconjugation, stability, electron delocalization, aromaticity (bonding character in organic molecules) are underway. The first stable disubstituted linear 14π annulene was recently prepared [J. Am. Chem. Soc. (2003)] in this series with the tandem Pauson-Khand reaction as the key step to generate molecular complexity.

G. We have in progress two projects directed toward the preparation of molecules which may house a planar four coordinate carbon atom. Initial targets are shown below. The Weiss reaction continues to be important in this work.

Rahman 7/17/2016